
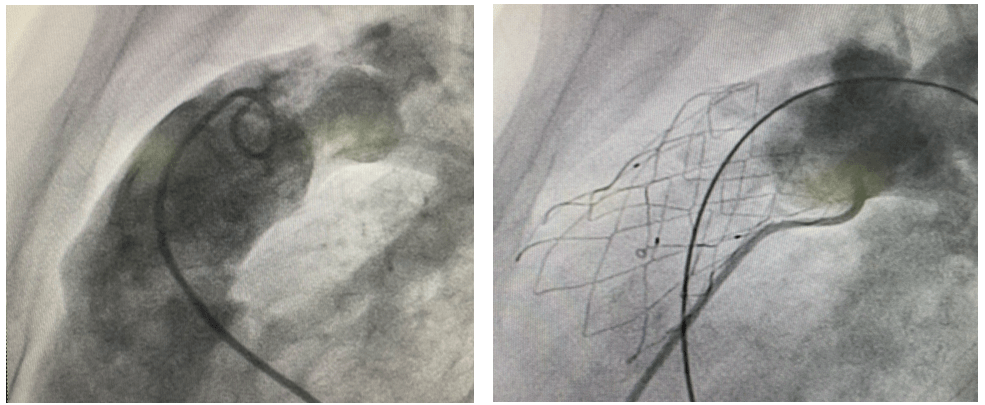
在那处于医疗领域特定范畴的消化内科手术这一过程当中,发挥着根本作用的扮演角色之物是胃底折叠手术所运用的钉合器,医生凭借内镜这一工具将它以较为轻柔的方式缓缓送至胃底部位,仅需“咔嚓”几声这样的操作过程,就能达成胃底与食管之间被牢牢钉合的效果,进而对胃食管反流所带来的困扰起到有效的缓解作用,不过,要想让此类有着特定功能用途的产品实现合规上市这样的目标,绝对不是一件轻而易举的事情,整个需要历经的注册流程,宛如身处一座无法瞧见出口究竟在何处的迷宫一般,每迈出的一步都务必以谨慎又坚定的姿态去推进,只有顺利通过药监部门一层又一层严格的把关审核之后,才能够迎来象征最终成功的那一束光亮。
")
需弄清楚的是,我们必须以极为清晰的方式去对产品的“身份”予以界定;依据自2020年7月开始直至2021年12月期间所发布的分类界定汇总信息来看,那用于胃底折叠手术的钉合器被弄清楚无误地划归到Ⅱ类医疗器械范畴,其具体条目乃是处于第193条之处;而这所意味的则是,与Ⅰ类产品相比较的话,它的注册之路就变得更为充满曲折,所需提交的相关材料也就变得更为繁杂,审查时所投射出的目光也更为锐利;企业方面不但要针对产品技术要求、风险研究报告以及检验报告等一系列文件做好充足准备,而且还要去直面技术审评这个环节,甚至要去应对质量体系的全方位核查工作;整个过程显得漫长并且体现出出环环相扣的状态,犹如行走在独木桥上一般,稍微出现一点疏忽,便极有可能在某一处出现“卡住”的状况,进而让前期为之付出的所有努力全然付诸东流。
步骤一:弄清楚产品分类与基本要求
作为Ⅱ类医疗器械的胃底折叠手术用钉合器,因其属于中风险产品因此注册流程相较于Ⅰ类产品而言必然更为复杂,企业需依照诸如2020年7月至2021年12月汇总中第193条建议这类《医疗器械分类目录》及相关分类界定通知,以准确判断该产品类别归属,此步看似简单却实则牵一发而动全身,盖因类别不但决定后续注册路径,而且影响所需资料深度与审评严格程度,一旦分类有误则申请很可能被退回,致使数月心血或许就此搁浅。
而且我们不仅要清晰地对产品那涵盖结构组成(像是钉合器主体以及包括钉仓、吻合钉与其他附件等方面)、灭菌方式(比如需考虑是否以无菌状态提供以及是不是一次性使用这般情况)、适用范围(应用于将胃底与食管开展钉合的胃底折叠手术方面)还有工作原理(像在超声引导之下开展定位并实施击发钉合操作这类情况)等多维度“样貌”加以勾勒,还需明白这些看似基础的信息在注册全程中实则扮演着贯穿始终的根本角色,且从起始阶段便务必确保其能精准无误且保持一致性,哪怕一丝模糊都绝不容许出现。
步骤二:准备技术文档与检测报告
在整个注册申请里存在着最为根本且繁杂到极点的一步,这一步需要准备数量颇多且种类繁杂同时得严格依照药监部门格式与内容要求的文件,像产品技术要求这类根本文件以及风险研究报告、产品说明书与标签样稿、检验报告与其他相关证明材料通常都被涵盖于其中。
在需将产品的各项性能指标诸如钉合强度、组织对合效果、击发力这类以及安全指标比如生物相容性、无菌性能还有与之对应的检验方法必须逐一详细列明的产品技术要求当中,产品在正常使用以及可预见误用等情形下有可能出现的全部风险需被风险研究报告开展系统评估,并且相应的控制策略也得被制定出来。
必须由那具备着国家所认可资质的医疗器械检测机构来出具检验报告这一点需弄清楚,尤其针对Ⅱ类产品而言,企业自行出具报告是被禁止的,而应委托有着相关资格的检测机构,依据企业所提交的产品技术要求去展开检测,进而出具正式报告;整个过程常常会耗时较长,期间检测机构有可能指出修改意见,在此种状况下需要企业积极予以配合,以便对产品或者技术文件做出相应调整,不过这其中所涉及到的各环节之间的复杂关系以及潜在影响因素,是难以简单说清且可能随时出现变化的。
步骤三:临床评价路径
并非全部的医疗器械皆务必凭借全新开展的临床试验用以对其自身的安全有效性加以证明,就像是胃底折叠手术当中所使用的钉合器这类已具备相对成熟特性的产品而言,在通常情况下更为常见且广泛应用的做法是选用“临床评价”这样的路径——那便是借由与已然于中国境内以合法方式获得上市资格的同类产品展开比较,进而针对本产品的安全有效性来实施论证的工作。
这所要求于企业的是需广泛去开展对文献资料以及临床数据的搜集工作,且要针对本产品同对照产品在诸如结构组成、性能指标、适用范围还有工作原理等等这些方面的异同情况展开详细比较,重点在于要对差异部分开展阐释,考量其是否引入全新风险或者是否对产品的安全有效性形成影响。而若这种比较无法做到充分证明,又或者产品属于创新类别且缺乏适宜对照之时,临床试验便可能被要求启动,需知晓临床试验有着周期长、成本高的特点,其涉及从伦理审查、临床备案、患者入组到数据收集以及报告撰写一系列极为复杂的流程。
步骤四:提交注册与应对审评
当所有的技术资料以及各类检测报告连同需要认真考量的临床评价文件,按照既定标准全部准备得妥妥当当之后,对于境内第二类医疗器械而言的相关企业,其注册申请可被提交至所在地的省级药品监督管理部门这个特定主体之处。
药监部门在受理之后,所采取的步骤为先行展开针对资料是否齐全以及是否符合基本要求的形式审查工作,待此审查完成确认满足条件后,资料将会被转交到技术审评机构以开展根本的实质性审评环节,在这一根本环节里,审评专家会对资料开展逐项的详细审阅,并从全面的角度去评估产品的安全性与有效性有没有得到充分的验证,虽然法定审评时限规定为60个工作日,然而在实际操作过程中,却常常因为出现需要“补充资料”也就是发补的情况致使时间延长,而企业则必须在接收到发补通知之后于规定的通常时长大约一年的时间范围内,完成资料的补充并重新予以提交。
在历经技术审评得以顺利通过这一环节之后,生产企业的质量管理体系便会成为药监部门所着重关注并即将开展现场核查工作的对象,此核查行动的重要目的就是因为能够切实确保具备如此能力的企业,即持续稳定生产出合格产品的能力的企业,在后续的生产过程中一直保持相应水平。
步骤五:体系核查与获证
医疗器械注册过程中,起着根本作用的质量管理体系核查这一根本的环节里,届时药监局会派员深入被核查企业的生产现场,他们会依据《医疗器械生产质量管理规范》等一系列法规,对包括企业组织机构、人员培训,还有厂房设施、设备管理,以及物料控制、生产过程,甚至质量控制与文件记录管理等等在内的各个环节,展开既全面又细致入微的检查;而企业自身则必须提前形成起并且确保能有效运行符合相关要求的质量管理体系,做好各类充分准备以迎接核查;值得注意的是,要是在这个体系核查当中被发现存在严重不符合项的情况,那么极有可能直接对注册审批进度造成影响,甚至在一定程度上决定着最终能否获得批准。
只有当技术审评伴随着体系核查二者均顺利通过这种情况出现之后,在法定期限内,也就是通常来说在技术审评结束过后的20个工作日之内,药监部门才会就医疗器械企业是否准予注册这一根本事项作出相应决定,对于那些被判定符合所有既定要求的企业,医疗器械注册证将会由药监部门予以发表,而只有成功拿到这根本如同“通行证”一般的医疗器械注册证,产品方可合法依规地开展生产活动以及实现上市并展开销售等一系列商业运作行为。
步骤六 上市后监督与延续注册
当成功拿到注册证之时,其实这远远不能视为终点,而应将其当作一段崭新的、充满着各类复杂责任的起点之处,这就要求企业必须以极为严谨且切实可行的方式,来履行在产品顺利上市之后所面临的各项复杂义务,这其中包括了需形成一套行之有效的不良事件监测与详细报告制度,通过系统且全面地去收集、有条理地记录并且深入细致地研究产品在上市之后于实际使用过程当中所出现的各类情况,倘若一旦发现任何哪怕仅有一丝可疑迹象的不良事件,那么无论该事件看起来多么微小,都必须以最快速度、最妥善方式向具有监管职责的药监部门作出及时报告。
当产品于设计层面、材料使用状况、工艺操作流程以及适用范围等多领域出现那类对安全有效性造成影响的变更情形之时,依据规定,企业被要求必须向负责监管的药监部门及时提交与之对应的变更注册申请或者备案申请,并且只有在获得批准之后,相关变更方可着手实施。另外不得不提及的是,通常而言医疗器械注册证是设有一定有效期的,比如说以5年为例。在此基础上,企业应当在注册证即将到期之前的某一段特定时间(就像6个月那样)就主动启动延续注册的相关程序,向密切相关部门提交产品自上市之后所逐步积累起来的安全有效性数据,同时提交能够证明其符合当下最新法规要求的各类证明文件,以此来完成整个延续注册申请工作。
当您脑海之中开始萌生出针对于胃底折叠手术用钉合器开展注册这样一种计划的时刻,那么被强烈建议的一种做法乃是去咨询诸如具备如飞速度那般专业能力的CRO此类的专业从事于咨询相关事务的机构,通过此方式进而获取得到在内容方面更为详尽、全面且具有指导性意义以及全方位特性的相关指导与服务,尽管这样的过程可能会因种种未知因素而变得极为复杂,或许还需要考虑众多并不明晰的方面,然而这一途径被广泛认为是在当前情境下相对可靠之选。

 1362
1362

